

Abstract
Introduction: Epigenetic dysregulation plays a critical role in hematologic tumorigenesis and cancer progression. Histone-lysine N-methyltransferase 2D (KMT2D), also known as MLL4 (mixed-lineage leukemia 4), belongs to a family of mammalian histone H3 lysine 4 (H3K4) methyltransferases, which is amongst the most frequently mutated epigenetic genes in human cancers. It was reported that deletion of KMT2D inhibits MLL1-fusion-induced acute myeloid leukemia (Santos et al. 2014). However, in human cancers, the vast majority of the mutations in KMT2D is heterozygous deletion. Recent studies have revealed the role of KMT2D in lymphomagenesis, but the potential tumor suppressor function of KMT2D in leukemogenesis remains largely unclear. In addition, KMT2D was found to exist within a macromolecular complex named COMPASS (complex of proteins associated with Set1). KMT2C is another component of the complex, and was validated as another haploinsufficient tumor suppressor gene in acute myeloid leukemia (Chen et al. 2014). Mutations in KMT2D and KMT2C frequently co-occur in hematologic malignancies. Thus, we sought to first examine the role and molecular mechanism of KMT2D in acute leukemia, and then investigate whether co-mutated gene KMT2D and KMT2C have cooperative function in malignancies initiation and progression.
Methods: Here we utilized RNAi approach to knockdown the expression of Kmt2d, and an approximate 50% reduction in gene dosage was validated by quantitative RT-PCR. We introduced two independent short-hairpin RNAs (shRNAs) targeting Kmt2d and a shRNA targeting Nf1 into p53 null hematopoietic stem and progenitor cells (hereafter referred to as shKmt2d;shNf1;p53-/- cells), and transplanted them into sublethally irradiated mice to model the impact of haploinsufficient tumor suppressors. The recipient mice were then monitored for the emergence of diseases by complete blood count as well as overall survival. Bone marrow was harvested and analyzed by flow cytometry after sacrifice. Histological analyses of blood smear, liver, spleen and bone marrow were conducted to accurately diagnose the disease. Besides, we established Kmt2d heterozygous and homozygous conditional knockout mouse models (Kmt2d fl/+; Mx1-Cre, Kmt2d fl/fl; Mx1-Cre) to further investigate the respective role of haploinsufficiency and deletion of KMT2D in acute leukemia. To study the cooperative function of KMT2D and KMT2C in leukemogenesis, two genes were co-suppressed by shRNAs in the same aforementioned genetic background (shNf1; p53-/-). Transplantation-based mouse modeling approach was used and the recipient mice were monitored for evidence of hematologic diseases.
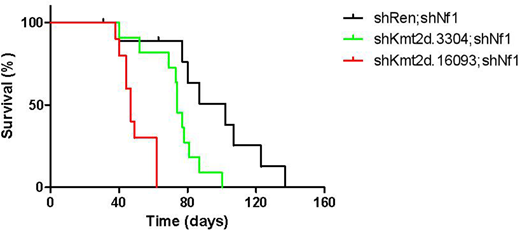
Results: A significant increase in peripheral white blood cell counts, anemia and thrombocytopenia were noticed in recipients of shKmt2d;shNf1;p53-/- cells over the same period after 4 weeks post-transplantation. Massive hepatomegaly and splenomegaly were displayed in those mice and a dramatic reduction in survival was observed (shKmt2d-1: median survival = 47 days, p = 0.0004 versus shRen; and shKmt2d-2: median survival = 74 days, p = 0.0127 versus shRen). Flow cytometry of cells from bone marrow of sacrificed mice showed a substantial enrichment of doule-positve cells (shRNAs targeting Kmt2d were linked to GFP and those targeting Nf1 were linked to mCherry) as compared to pre-injected, which were filled with leukemic cells that expressed myeloid/lymphoid lineage markers. Pathological analyses demonstrated the onset of acute myeloid/lymphoid leukemia. In Kmt2d and Kmt2c double-knockdown animal assay, the onset of acute myeloid leukemia was promoted compared with single-knockdown groups. (shKmt2d;shKmt2c: median survival = 48.5 days, p < 0.0001 versus shKmt2d; p = 0.0013 versus shKmt2c).
Conclusions: KMT2D is a haploinsufficient tumor suppressor in acute leukemia. KMT2D deficiency cooperates with KMT2C to promote the development and progress of acute leukemia. Further understandings of the molecular mechanism of KMT2D as a haploinsufficient tumor suppressor in acute leukemia and the cooperative function of KMT2D and KMT2C in leukemogenesis are demanded, which can provide insights into developing novel therapeutic targets.
No relevant conflicts of interest to declare.

